

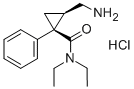
CAS号:101152-94-7
|
英文名称:(1R,2S)-rel-2-(Aminomethyl)-N,N-diethyl-1-phenylcyclopropanecarboxamide hydrochloride
分子式
C15H23ClN2O
分子量
283
EINECS号
620-477-4
MDL
MFCD00901293
Smiles
InChIKey
盐酸米那普仑化学百科
基本信息
| 中文名称 | 盐酸米那普仑 |
| 英文名称 | (1R,2S)-rel-2-(Aminomethyl)-N,N-diethyl-1-phenylcyclopropanecarboxamide hydrochloride |
| CAS号 | 101152-94-7 |
| 分子式 | C15H23ClN2O |
| 分子量 | 282.81 |
| EINECS号 | 620-477-4 |
物化性质
| 熔点 | 179-1810C |
| 闪点 | 9℃ |
| 溶解度 | H2O: 19 mg/mL |
| 形态 | 固体 |
| 颜色 | 白色 |
| 默克索引编号 | 14,6194 |
安全信息
| 危险品标志 | Xn,T,F |
| 危险类别码 | 22-39/23/24/25-23/24/25-11 |
| 安全说明 | 7-16-36/37-45 |
| 危险品运输编号 | UN 2811 6.1/PG 3 |
| WGK Germany | 3 |
| RTECS号 | GZ1014010 |
| 海关编码 | 29242990 |
| 危险等级 | 6.1 |
| 包装类别 | III |
| 毒性 | LD50 orally in mice: 237 mg/kg (Bonnaud) |
生产及用途
1)盐酸米那普仑是不可能涉及临床上有意义的药代动力学药物相互作用;
2)盐酸米那普仑与其它药物可能发生药效学相互作用 米那普仑的中枢疼痛抑制作用确切作用机制和在人中改善纤维肌痛症状的能力尚未知。临床前研究曾显示米那普仑是一种神经元去甲肾上腺素和5-羟色胺再摄取强抑制剂;在体外米那普仑抑制去甲肾上腺素摄取 比5-羟色胺强度约更高3-倍,对多巴胺或其它神经递质的摄取无直接影响。米那普仑在体外对5-羟色胺能(5-HT1-7),α-和β-肾上腺能,毒蕈硷(M1-5),组胺(H1-4),多巴胺(D1-5),鸦片,苯二氮卓,和γ-氨基丁酸(GABA)受体无明显亲和力。在这些受体处的药理学活性被假设是伴随各种抗胆碱能,镇静,和心血管效应与其它精神病治疗药物所见。米那普仑对Ca2+,K+,Na+和Cl– 通道无明显亲和力而且不抑制人单胺氧化酶(MAO-A和MAO-B)或胆碱酯酶活性。
Milnacipran HCl能抑制去甲肾上腺素转运蛋白(NET)和五羟色胺转运蛋白(SERT),其IC50分别是77 nM和420 Nm
2)盐酸米那普仑与其它药物可能发生药效学相互作用 米那普仑的中枢疼痛抑制作用确切作用机制和在人中改善纤维肌痛症状的能力尚未知。临床前研究曾显示米那普仑是一种神经元去甲肾上腺素和5-羟色胺再摄取强抑制剂;在体外米那普仑抑制去甲肾上腺素摄取 比5-羟色胺强度约更高3-倍,对多巴胺或其它神经递质的摄取无直接影响。米那普仑在体外对5-羟色胺能(5-HT1-7),α-和β-肾上腺能,毒蕈硷(M1-5),组胺(H1-4),多巴胺(D1-5),鸦片,苯二氮卓,和γ-氨基丁酸(GABA)受体无明显亲和力。在这些受体处的药理学活性被假设是伴随各种抗胆碱能,镇静,和心血管效应与其它精神病治疗药物所见。米那普仑对Ca2+,K+,Na+和Cl– 通道无明显亲和力而且不抑制人单胺氧化酶(MAO-A和MAO-B)或胆碱酯酶活性。
Milnacipran HCl能抑制去甲肾上腺素转运蛋白(NET)和五羟色胺转运蛋白(SERT),其IC50分别是77 nM和420 Nm
| Target | Value |
| norepinephrine transporter (NET) | 77 nM |
| norepinephrine transporter (SERT) | 420 nM |
Milnacipran主要以本体和葡萄糖醛酸苷的形式通过尿液排出(>80%),仅有少部分(<10%)通过CYP3A4酶催化的N端脱乙基作用代谢掉 Milnacipran在高浓度时能抑制某一配体离子通道(LGIC)受体,包括NMDA, 5-HT3A 和nACh受体,其IC50分别是58.4 μM, 185μM, 14.3μM。
Milnacipran(10和30 mg/kg, 口服)能增加大鼠前额皮质中细胞外的5-HT和 NA的水平,这种影响效应与剂量相关。在抑郁症动物行为模型中,Milnacipran (30和60 mg/kg, 口服)能显著降低大鼠的强迫性游泳实验中的静止时间。在焦虑症动物行为模型中,Milnacipran (30和60 mg/kg, 口服)也能显著降低大鼠条件性恐惧压力测试实验中不动时间。 Milnacipran (<40 mg/kg 腹腔注射)可以增加自由活动豚鼠下丘脑中细胞外的NA和5-HT的水平,这种作用是剂量依赖的。在自由活动豚鼠下丘脑中,分别用10 mg/kg和40 mg/kg Milnacipran处理,分别能减少57%和47%的NA代谢产物MHPG。化学性质
白色结晶性粉末。上下游产品
上游产品共计:0个
下游产品共计:0个
相关产品
-
CAS号:138982-67-9
齐拉西酮盐酸盐一水合物 , 一种有效的dopamine 和 serotonin (5-HT) receptor的拮抗剂 -
CAS号:85650-52-8
米尔塔扎平 , 5-HT受体抑制剂 -
CAS号:131740-09-5
夫拉平度盐酸盐 -
CAS号:871700-17-3
曲美替尼 , 一种MEK1/2 抑制剂 -
CAS号:54910-89-3
氟西汀 -
CAS号:21535-47-7
盐酸米安色林 -
CAS号:101152-94-7
盐酸米那普仑 -
CAS号:3092-17-9
盐酸米多君 -
CAS号:379231-04-6
萨拉卡蒂尼(AZD0530) -
CAS号:13614-98-7
盐酸米诺环素 -
CAS号:59277-89-3
阿昔洛韦 (LC) -
CAS号:92623-85-3
米那普仑 -
CAS号:606143-52-6
司美替尼 -
CAS号:116539-59-4
度洛西汀 -
CAS号:179324-69-7
硼替佐米 -
CAS号:67915-31-5
曲康唑 -
CAS号:82034-46-6
依碳氯替泼诺 -
CAS号:138982-67-9
齐拉西酮盐酸盐一水合物 , 一种有效的dopamine 和 serotonin (5-HT) receptor的拮抗剂 -
CAS号:85650-52-8
米尔塔扎平 , 5-HT受体抑制剂 -
CAS号:131740-09-5
夫拉平度盐酸盐 -
CAS号:871700-17-3
曲美替尼 , 一种MEK1/2 抑制剂 -
CAS号:54910-89-3
氟西汀 -
CAS号:21535-47-7
盐酸米安色林 -
CAS号:101152-94-7
盐酸米那普仑 -
CAS号:3092-17-9
盐酸米多君 -
CAS号:379231-04-6
萨拉卡蒂尼(AZD0530) -
CAS号:13614-98-7
盐酸米诺环素 -
CAS号:59277-89-3
阿昔洛韦 (LC) -
CAS号:92623-85-3
米那普仑 -
CAS号:606143-52-6
司美替尼 -
CAS号:116539-59-4
度洛西汀 -
CAS号:179324-69-7
硼替佐米 -
CAS号:67915-31-5
曲康唑 -
CAS号:82034-46-6
依碳氯替泼诺
产品供应商
